

Mécanismes et bases génétiques
Le fonctionnement du corps humain : de la cellule à l’ADN
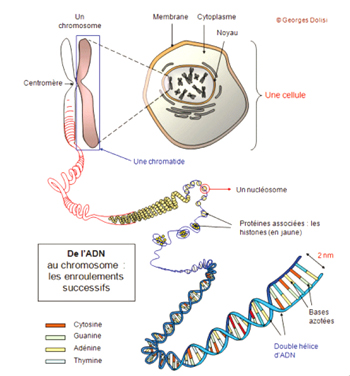 Notre corps est fait de millions de cellules et nous possédons normalement 46 chromosomes dans chacune de nos cellules : un jeu de 23 chromosomes hérité de notre mère, et un jeu de 23 chromosomes hérité de notre père soit 23 paires. Les chromosomes, composés d’ADN, contiennent les gènes qui définissent les caractéristiques génétiques d’un individu. Nous héritons donc de deux copies de chaque gène: une copie de chacun de nos parents.
Notre corps est fait de millions de cellules et nous possédons normalement 46 chromosomes dans chacune de nos cellules : un jeu de 23 chromosomes hérité de notre mère, et un jeu de 23 chromosomes hérité de notre père soit 23 paires. Les chromosomes, composés d’ADN, contiennent les gènes qui définissent les caractéristiques génétiques d’un individu. Nous héritons donc de deux copies de chaque gène: une copie de chacun de nos parents.
L’ADN est le support de l’information génétique qui oriente et dicte la construction et le fonctionnement du corps humain. Les gènes sont un fragment d’ADN.
La perte de matériel génétique
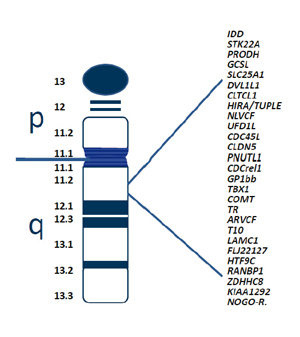 Dans le cas de la Délétion 22q11, un fragment d’ADN contenant plusieurs gènes est perdu sur l’un des deux chromosomes 22 au moment de la formation des gamètes (ovocytes ou spermatozoïdes) avant la fécondation. Il se produit une erreur irréversible d’écriture lors de la copie de l’information génétique au niveau de la séquence d’ADN.
Dans le cas de la Délétion 22q11, un fragment d’ADN contenant plusieurs gènes est perdu sur l’un des deux chromosomes 22 au moment de la formation des gamètes (ovocytes ou spermatozoïdes) avant la fécondation. Il se produit une erreur irréversible d’écriture lors de la copie de l’information génétique au niveau de la séquence d’ADN.
Nous sommes en présence d’une mutation génétique car l’absence de certains gènes sur le chromosome 22 va modifier le fonctionnement du corps humain en perturbant plus ou moins fortement le développement de l’embryon. En effet, si une seule copie du gène est présente, son expression peut se retrouver altérée.
Par exemple
- le gène TBX1 est impliqué dans la formation de la paroi des vaisseaux sanguins. Il contrôle également l’expression de nombreux autres gènes qui sont importants dans le développement embryonnaire,
- le gène PRODH régule la proline (acide aminé dont se servent les organismes vivants pour assembler les protéines) et joue un rôle dans les symptômes psychotiques et cognitifs,
- le gène COMT joue un rôle de régulation de la dopamine et affecte les performances cognitives.
Les effets de la délétion sur l’embryon
La délétion des gènes situés sur le locus 22q11 va perturber le mouvement ou la différentiation des cellules de la crête neurale, et par la suite affecter leur développement. Ceci entraîne chez les personnes porteuses du syndrome, principalement :
- des malformations du massif crânio-facial, (face et noyaux moteurs des nerfs crâniens),
- des anomalies de la commande motrice de la succion et de la déglutition,
- des malformations cardio-vasculaires,
- une hypoplasie thymique (insuffisance du développement du thymus).
On constate cependant que ces malformations sont variables d’une personne à l’autre sans qu’il soit possible pour le moment de donner des explications précises. Il apparaît que la perte de matériel génétique soit mieux compensée chez certains individus.
La transmission
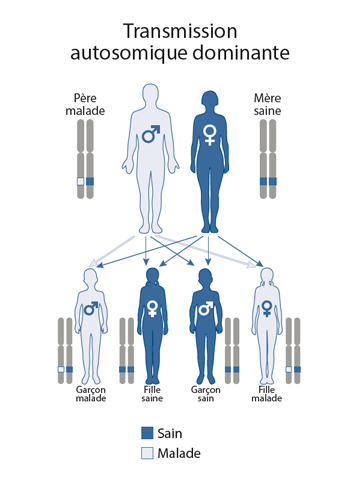 Dans 80 à 90% des cas, la Délétion 22q11 apparaît de novo, c’est-à-dire accidentellement ; aucun des deux parents n’est porteur de l’anomalie génétique. Dans 10 % à 20 % des cas seulement, un des deux parents est porteur.
Dans 80 à 90% des cas, la Délétion 22q11 apparaît de novo, c’est-à-dire accidentellement ; aucun des deux parents n’est porteur de l’anomalie génétique. Dans 10 % à 20 % des cas seulement, un des deux parents est porteur.
Pour les personnes porteuses de la Délétion 22q11 la transmission aux enfants est dite : « autosomique dominant ».
Autosomique veut dire que les filles comme les garçons peuvent transmettre la délétion car les gènes manquants ne se situent pas sur le chromosome sexuel.
Dominant : les signes de la maladie sont présents dès lors que l’anomalie est présente sur l’un des deux chromosomes 22.
Dans ce mode de transmission, l’un des parents porte une copie délétée du chromosome 22, c’est-à-dire sur laquelle il manque une vingtaine de gènes. Le parent atteint par le syndrome peut, lors de la méiose, transmettre la copie délétée ou la copie saine. La probabilité de transmettre la délétion à son enfant est de 50%.
Bon à savoir
Pour les mutations de novo, le risque théorique qu’un frère ou une sœur soit touché lorsque les parents sont indemnes est nul. Dans les faits, le risque pratique est de 1 à 2% : la plupart du temps, la mutation est déjà présente soit dans le spermatozoïde soit dans l’ovule qui a été à l’origine de la personne malade. Or ce spermatozoïde ou cet ovule sont issus de la division des cellules souches qui sont situées dans les testicules ou les ovaires des parents. Lorsque la mutation s’est produite dans une cellule souche elle sera présente non pas dans un spermatozoïde ni dans un ovule mais dans plusieurs. C’est ce qu’on appelle une Mosaïque Germinale
Le nombre d’anomalies et la gravité de chacune d’entre elles ne sont pas systématiquement identiques entre un parent et son enfant, tous deux porteurs de la microdélétion. On observe une grande variabilité des symptômes.
Par exemple, lorsqu’un des parents est atteint et transmet à son enfant la Délétion 22q11 :
- si ce parent a une malformation cardiaque, rien ne laisse prédire que l’enfant aura aussi une malformation cardiaque,
- si ce parent n’a pas de malformation cardiaque, rien ne certifie que l’enfant n’aura pas de malformation cardiaque.
Il est recommandé que les parents des personnes atteintes soient testés, car il arrive qu’un des parents soit si légèrement atteint qu’avant d’être lui même testé il ignore être porteur de la délétion.
Le nombre d’anomalies et la gravité de chacune d’entre elles ne sont pas systématiquement identiques entre un parent et son enfant, tous deux porteurs de la délétion. On observe une grande variabilité des symptômes.
Par exemple, lorsqu’un des parents est atteint et transmet à son enfant la Délétion 22q11 :
si ce parent a une malformation cardiaque, rien ne laisse prédire que l’enfant aura aussi une malformation cardiaque,
si ce parent n’a pas de malformation cardiaque, rien ne certifie que l’enfant n’aura pas de malformation cardiaque.
Il est recommandé que les parents des personnes atteintes soient testés, car il arrive qu’un des parents soit si légèrement atteint qu’avant d’être lui même testé il ignore être porteur de la délétion.